Fibrosi Cistica: MUC5B e soluzione ipertonica

Nuovo studio sulla fibrosi cistica:
maturazione postsecretoria difettosa della mucina MUC5B nelle vie respiratorie
Le persone affette da fibrosi cistica (FC) soffrono di continue infezioni ai polmoni a causa di un muco così denso e viscoso da non riuscire a catturare da batteri, virus e altri agenti patogeni. Le cause della natura insolita di questo muco nella fibrosi cistica non sono ancora chiare ma i ricercatori della Scuola di Medicina della UNC hanno trovato un indizio importante.
La loro pubblicazione sul JCI Insight ci dice che le proteine del muco, che donano la caratteristica gelatinosità, non vengono prodotte normalmente nelle vie respiratorie di chi è affetto da fibrosi cistica.
“Nelle persone in salute, dopo che le cellule in superficie hanno prodotto il muco, le proteine passano da una formazione più compatta a una più lineare e aperta,” ci racconta l’autore e ricercatore Mehmet Kesimer, professore associato di Medicina Patologica e di Laboratorio e membro dell’Istituto di Pneumologia di Marsico dell’UNC, “Abbiamo scoperto che nei casi di fibrosi cistica questo processo di disposizione è difettoso nell’epitelio delle vie respiratorie”.
La fibrosi cistica
La fibrosi cistica è una malattia genetica rara che affligge circa 70.000 persone nel mondo. Si presenta quando un individuo ha due copie difettose del gene CFTR; ciò scatena la produzione della proteina CFTR. Nella persona in salute, la CFTR permette al cloruro e agli altri ioni di uscire dalle cellule, incluse quelle epiteliali che permeano le vie respiratorie tra i polmoni e la gola.
“Questi ioni di cloruro non fanno altro che trattenere l’acqua fuori dalle cellule. Abbiamo bisogno che ci sia dell’acqua proprio qui. Quando il flusso di cloruro si riduce, la superficie delle cellule si disidrata”. È quindi la mancanza di acqua che aumenta la viscosità del muco.
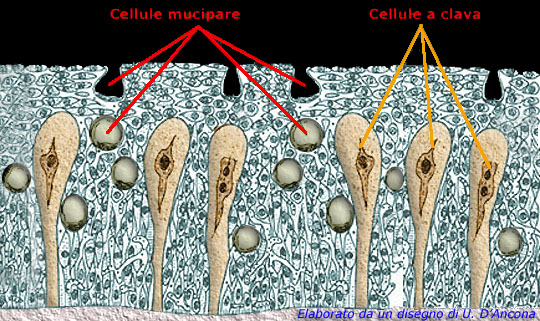
Cellule Mucipare e Cellule a Clava,
qui viene prodotta la MUCB5
La studio
Normalmente le proteine mucine, in particolare la MUC5B, si dispongono in pochi minuti o al massimo in poche ore, mentre nei casi di fibrosi cistica rimangono compatte. La microscopia elettronica ha confermato la struttura abnorme e densa della MUCB5 secreta dalle cellule di chi vive con la FC.
Questo studio suggerisce che la mancanza di uno strato di acqua sulle cellule provoca questa struttura insolita della MUCB5 e conferma che la reidratazione delle vie respiratorie ripristina la natura fluida del muco e il benessere del paziente.
Le irrigazioni con soluzione ipertonica
Altri studi clinici avevano già dimostrato che le irrigazioni di soluzione salina ipertonica – acqua sterile salata, che favorisce il ripristino dell’equilibrio ionico normale e quindi la reidratazione delle vie respiratorie – assottiglia il muco e rallenta il declino delle funzioni polmonari.
Inoltre i ricercatori mirano ai legami chimici all’interno delle mucine. Questo approccio potrebbe portare allo scioglimento di queste strutture dense e grandi di MUCB5 in piccoli frammenti.
Adesso Kesimer e i suoi colleghi hanno prove a sufficienza per dire che le mucine sono ciò che conta maggiormente e che la chiave è nello strato acquoso su cui il muco si muove.
Defective postsecretory maturation of MUC5B mucin in cystic fibrosis airways, Lubna H. Abdullah, Jessica R. Evans, T. Tiffany Wang, Amina A. Ford, Alexander M. Makhov, Kristine Nguyen, Raymond D. Coakley, Jack D. Griffith, C. William Davis, Stephen T. Ballard, and Mehmet Kesimer, JCI Insight, doi: 10.1172/jci.insight.89752, pubblicato il 23 Marzo 2017.
Trad. it a cura dell’autore, fonte: medicalnewstoday.com


