Cystic Fibrosis: MUC5B and Hypertonic Solution

New study on cystic fibrosis:
Defective postsecretory maturation of MUC5B mucin in the respiratory tract
People with cystic fibrosis (CF) suffer from constant lung infections because their mucus is so thick and sticky that it cannot trap bacteria, viruses, and other pathogens. The causes of the unusual nature of this mucus in cystic fibrosis are still unclear, but researchers at the UNC School of Medicine have found an important clue.
Their publication on JCI Insight tells us that the proteins in mucus, which give it its characteristic gelatinous texture, are not produced normally in the respiratory tract of people with cystic fibrosis.
"In healthy people, after the cells on the surface have produced mucus, the proteins change from a more compact formation to a more linear and open one," says author and researcher Mehmet Kesimer, associate professor of Pathological and Laboratory Medicine and member of theUNC Marsico Institute of Pulmonary Medicine. "We have discovered that in cases of cystic fibrosis, this arrangement process is defective in the epithelium of the respiratory tract."
Cystic fibrosis
Cystic fibrosis is a rare genetic disorder that affects approximately 70,000 people worldwide. It occurs when an individual has two defective copies of the CFTR gene, which triggers the production of the CFTR protein. In healthy individuals, CFTR allows chloride and other ions to exit cells, including epithelial cells that line the airways between the lungs and throat.
"These chloride ions simply keep water out of the cells. We need water right here. When the flow of chloride is reduced, the surface of the cells becomes dehydrated." It is therefore the lack of water that increases the viscosity of the mucus.
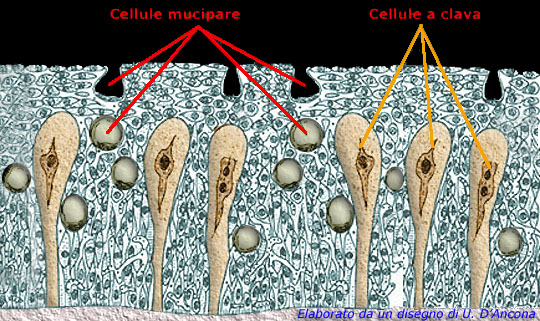
Mucipare Cells and Clava Cells,
where MUCB5 is produced
The study
Normally, mucin proteins, particularly MUC5B, arrange themselves within a few minutes or at most a few hours, whereas in cases of cystic fibrosis they remain compact. Electron microscopy has confirmed the abnormal, dense structure of MUCB5 secreted by the cells of people living with CF.
This study suggests that the lack of a layer of water on the cells causes this unusual structure of MUCB5 and confirms that rehydration of the airways restores the fluid nature of the mucus and the well-being of the patient.
Irrigation with hypertonic solution
Other clinical studies had already demonstrated that irrigation with hypertonic saline solution – sterile salt water, which promotes the restoration of normal ionic balance and thus the rehydration of the respiratory tract – thins mucus and slows the decline in lung function.
In addition, researchers are targeting the chemical bonds within mucins. This approach could lead to the dissolution of these dense, large MUCB5 structures into small fragments.
Now Kesimer and his colleagues have enough evidence to say that mucins are what matter most and that the key lies in the aqueous layer on which the mucus moves.
Defective postsecretory maturation of MUC5B mucin in cystic fibrosis airways, Lubna H. Abdullah, Jessica R. Evans, T. Tiffany Wang, Amina A. Ford, Alexander M. Makhov, Kristine Nguyen, Raymond D. Coakley, Jack D. Griffith, C. William Davis, Stephen T. Ballard, and Mehmet Kesimer, JCI Insight, doi: 10.1172/jci.insight.89752, published March 23, 2017.
Translated by the author, source: medicalnewstoday.com